Компьютерная геномика – новая эра. Дрозофилы с древним геном опровергли одну из классических теорий эволюции Прочитан не значит расшифрован
Ранее биологи изучали формы и функции организмов, переходя от изучения целого к изучению частей живого – органов и тканей. Современная биология изучает части – гены и молекулы, пытаясь воссоздать картину их функционирования в целом организме. Появилась новая наука – геномика . Объектом ее является совокупность всей генетической информации организма – геном. Работа генов определяет, какие белки синтезируются в клетке. Именно от разнообразия и активности белков зависят молекулярные процессы, обеспечивающие жизнь клетки и организма. Установление и характеристика полного набора белков данного организма относится к компетенции еще одного нового направления биологии – протеомики (от protein – белок).
В настоящее время число генов в геноме человека оценивается примерно в 30 тыс. Большинство генов в каждой клетке «молчит». К постоянно работающим во всех клетках относятся гены, кодирующие компоненты аппарата синтеза белка, РНК, ферменты, занимающиеся синтезом и починкой ДНК, ферменты системы обеспечения энергией и другие компоненты, необходимые для ведения «домашнего хозяйства» клетки. В геноме человека заведуют «домашним хозяйством» около одной пятой всех генов. Среди остальных есть гены, работающие только на определенных этапах развития организма, – например, те, которые кодируют эмбриональные белки или обеспечивают лактацию. Другие работают лишь в немногочисленных клетках. Так, способность воспринимать запахи связана с обонятельными рецепторами. Всего найдено около 1 тыс. генов, кодирующих рецептор, и в каждой клетке обонятельной луковицы работает только один ген. К удивлению исследователей, оказалось, что некоторые из этих генов работают и в сперматозоидах (предположительно, потому, что им надо как-то в темноте ориентироваться).
Можно сказать, что в каждой клетке «звучит» свой аккорд генов, определяя спектр синтезируемых на них видов РНК, кодируемых матричной РНК белков и, соответственно, свойства клетки. Набор активных генов различается в зависимости от типа ткани, периода развития организма, полученных внешних или внутренних сигналов.
В генетическом коде перед началом гена находятся промоторы – специальные регуляторные последовательности, которые включают и выключают гены в зависимости от полученных клеткой сигналов. Клетка прекрасно «понимает», когда и какой участок генетического текста надо считывать, реализуя записанную в нем информацию. Более 20 лет назад была расшифрована структура регуляторных участков в ДНК бактерий. С тех пор ученым удалось многое понять в работе генов.
Бурное развитие геномики связано, с одной стороны, с совершенствованием методов секвенирования ДНК, т.е. определения в ней последовательности (англ. sequence – последовательность) нуклеотидов. С другой стороны, лавинообразный рост объема накопленных данных вызвал развитие компьютерных технологий анализа информации, записанной в ДНК. Сейчас изучают уже не отдельные фрагменты ДНК, а целые геномы десятков бактерий, дрожжей, червя-нематоды, мухи дрозофилы.
Конечно же, наибольшие ожидания связаны с изучением генома человека, направленным на выявление нуклеотидных последовательностей всех генов человека, установление их функций и взаимодействий в норме и при нарушениях, приводящих к болезням.
Однако сам по себе нуклеотидный текст – только лишь исходный материал для дальнейшего анализа и выявления в нем генов. Необходимо понять, когда и какие фрагменты ДНК-текстов переписываются в набор молекул РНК, какие белки при этом синтезируются, как меняется при этом структура и функция клетки.
Кроме понимания фундаментальных законов жизни открытия геномики приносят заметную практическую пользу. Фармацевтические компании вкладывают огромные средства в геномные исследования. И не зря. Геномика уже многое дала медицине. А по предсказанию Френсиса Коллинза, возглавляющего американскую программу исследования генома человека, через 40 лет лечение самых различных недугов будет основано на использовании синтетических генных продуктов, которые будут изменять работу заболевших клеток и органов в нужном для выздоровления направлении.
Компьютерные эксперименты
В течение 100 лет изучение генов было основано на экспериментах in vivo (в живой клетке) и in vitro (в пробирке). Появившиеся в 1960-х гг. компьютеры были лишь вспомогательным средством для обработки и хранения данных. С конца 1980-х гг. началось создание баз данных, в которых хранится информация о миллионах последовательностей нуклеотидов в ДНК и РНК или аминокислот в белках. Компьютерный анализ превратился в самостоятельную область науки – биоинформатику . Исследования in silico , т.е. в компьютере, уже привели к расшифровке многих «слов» генетического текста – команд, записанных в ДНК и управляющих жизнью клетки.
Для такой расшифровки используют специально разработанные программы, например, для статистического анализа распределения нуклеотидов в ДНК. Напомним, что в генетическом алфавите всего четыре буквы – А (аденин), Т (тимин), Г (гуанин) и Ц (цитозин). Любители подсчета буковок выяснили, сколько может идти подряд букв А или как часто Г встречается после Ц в ДНК бактерий или человека.
В «осмысленных», т.е. кодирующих, участках ДНК эти сочетания подчиняются определенным правилам, тогда как в промежутках между генами, там, где ничего существенного в ДНК не записано, частота сочетаний нуклеотидов близка к случайной. Похоже на правила грамматики, которые мы учили в школе: «жи , ши пиши через и ». В словах русского языка буква «ы» после «ж» не встречается (разве что в тетрадях двоечников). Так же и в генетических текстах – в кодирующих участках некоторые сочетания нуклеотидов практически не встречаются, а распределение других сочетаний сильно отличается от случайного. Это видно при подсчете распределения триплетов, кодирующих аминокислоты, в генах излюбленного объекта генетиков – кишечной палочки (см. таблицу на с. 11). У кишечной палочки, так же как и у других организмов, всего имеется 64 триплета. Три из них – ТАА, ТАГ и ТГА – не кодируют аминокислоты, а являются сигналом окончания (терминации) синтеза белка.
Одна и та же последовательность ДНК может быть прочитана тремя способами со сдвигом на один нуклеотид. Способ чтения называется рамкой считывания . Понятно, что терминирующие триплеты не встречаются внутри гена в кодирующей рамке считывания (в других рамках они могут встречаться, но никому там не мешают – они не читаются, если молекулярная машина не собьется). Кодирующие триплеты распределены внутри гена по-разному. Триплет ЦТГ встречается примерно в 10 раз чаще, чем триплет ЦТА, хотя оба они кодируют одну и ту же аминокислоту – лейцин. В межгенных промежутках такие различия частот не наблюдаются.
Такие подсчеты, называемые статистическими методами анализа нуклеотидных последовательностей, позволяют распознать участки генома с определенными свойствами. Например, у бактерий большинство болезнетворных генов находится в так называемых островках патогенности, которые отличаются от остального генома по частоте встречаемости пар нуклеотидов А–Т и Г–Ц.
Где искать гены?
У бактерий выявить гены относительно легко. Во-первых, они занимают 80–90% бактериального генома, так что вероятность попасть в ген гораздо больше, чем промахнуться. Во-вторых, кодирующий участок бактериального гена – это непрерывная открытая рамка считывания, так что если ученому удалось найти в сплошной цепочке букв начало бактериального гена, то он будет читать его, как и клетка, триплетами до самого конца, пока не наткнется на стоп-кодон.
Гены высших организмов, в том числе и человека, искать намного труднее. У человека на участки, кодирующие белки, приходится только 5% генома. При этом кодирующие участки идут не сплошь, как у бактерий, а прерываются вставочными последовательностями – интронами, которые после синтеза матричной РНК из нее вырезаются. Ген может содержать до нескольких десятков кодирующих фрагментов – экзонов, чередующихся с интронами. К тому же в разных клетках могут использоваться разные сочетания экзонов одного и того же гена.
На основе статистического анализа можно с определенной долей вероятности установить, к какому участку генома относится исследуемый фрагмент. Подобно тому, как, включив телевизор и услышав слова «В отличие от обычного средства» или «Дешевле только даром», вы сразу поймете, что попали на рекламный ролик. Эти словосочетания в других передачах почти не встречаются. Так и определенные сочетания нуклеотидов указывают на принадлежность анализируемого куска генетического текста к интрону или экзону, кодирующему белок. Границы интронов и экзонов обозначены в ДНК специальным сочетанием нуклеотидов.
Без компьютерных биоинформационных технологий развитие геномных исследований было бы невозможным. Компьютерный поиск генов особенно важен для исследования генома человека, т.к. методы классической генетики имеют в этом случае ограниченное применение – ведь человек, в отличие от мух-дрозофил, не может быть объектом искусственного мутагенеза или иных генетических экспериментов.
Однако результаты экспериментов, поставленных на животных, могут быть применимы и к человеческому геному. Наиболее важные участки генома относительно мало изменяются в процессе эволюции, и их функции, установленные в экспериментах на мышах или мухах, оказываются такими же и у человека. Компьютерный анализ генетических текстов разных организмов позволяет выявить такие сходные участки.
Медленнее всего меняются участки, кодирующие белки. В наиболее важных белках отдельные участки сохранялись неизменными на протяжении миллиардов лет эволюции – от бактерий до человека. Это дает возможность находить гены при сравнении геномов отдаленно родственных видов. Таким анализом занимается сравнительная геномика. Ее методы используют для выявления родства отдельных генов, родства организмов, установления происхождения видов и более крупных таксонов.
Если сравнивать геномы человека и дрозофилы, то легко выявить гены, т.к. у неродственных организмов более заметна разница между значимыми (медленно меняющимися) и незначимыми участками. Но часть человеческих генов не похожа на мушиные, и выявить их при таком сравнении не удается. У нашей более близкой родственницы – мыши – почти такой же набор генов, как и у человека. Однако сохранение сходства в некодирующих областях создает трудности при выявлении генов сравнительными методами. т.е. выбирать организм для сравнения надо в зависимости от конкретной задачи.
А вот гены шимпанзе почти идентичны человеческим. Генетический текст шимпанзе отличается от нашего в среднем одним нуклеотидом из 300. Так что сравнение генома человека с обезьяньим нельзя использовать для выявления генов. Однако если гены уже известны, то существенные различия между человеком и шимпанзе скорее всего связаны именно с теми генами, которые делают нас людьми. Поэтому, как сообщил недавно журнал Science , в Германии начат проект «Геном шимпанзе».
Сравнительная геномика позволяет по известным функциям генов мухи или червя-нематоды предсказывать функции генов человека. А выявленные у человека гены, работа которых нарушена при тех или иных заболеваниях, могут быть изучены на других животных. Например, у человека найдены гены, мутации в которых приводят к болезни Альцгеймера – одной из форм старческого слабоумия. Оказалось, что изучать действие этих генов и искать способы лечения можно в экспериментах на мухах. Мутации в генах мухи приводят к изменениям в мушиных мозгах, очень сходным с молекулярными нарушениями, происходящими в мозге пациентов с болезнью Альцгеймера. У «слабоумных» мух нарушается способность к запоминанию. Ведется поиск генов мух, связанных с нарушениями памяти, и препаратов, способных замедлить развитие болезни, – сначала у мух, а потом, надо надеяться, и у людей. Важные для медицины и промышленности результаты дали исследования целых геномов бактерий. Уже полностью прочитаны геномы нескольких десятков бактерий. Среди них, кроме уже упоминавшейся кишечной палочки, возбудители социально значимых инфекций – туберкулеза, сифилиса, возбудители тифа, гастрита, некоторые промышленно важные бактерии. Практически все гены в изученных бактериальных геномах выявлены, для многих известны функции белкового продукта. По известным функциям белков проводят реконструкцию обмена веществ – метаболических путей бактерии. Реконструкция основных метаболических процессов организма по последовательности нуклеотидов его генома – одна из важнейших задач геномных исследований. Эту область исследований назвали труднопроизносимым словом метаболомика .
Анализ полной последовательности нуклеотидов генома микобактерии – возбудителя туберкулеза – показал, что у бактерии имеются жизненно важные для нее ферменты, отсутствующие у человека. Поиск лекарств, действующих именно на эти ферменты, обещает переворот в борьбе с инфекцией, уносящей миллионы человеческих жизней.
Перспективы биоинформатики
Экспериментальный поиск одного гена занимает недели и месяцы работы целой лаборатории. Компьютерные методы позволяют сделать это за считанные минуты, если просеквенирована ДНК организма и если есть хорошие алгоритмы поиска. Созданием таких алгоритмов и занимаются специалисты по биоинформатике. Различные программы используют для поиска генов, поиска регуляторных сигналов в ДНК, предсказания структуры и функций белка, его локализации в клетке, для реконструкции метаболизма. Реконструкция метаболических реакций, происходящих в разных клетках и тканях, будет одним из следствий расшифровки генетической информации человека.
Надо отметить, что российская биоинформатика не отстает, а зачастую и опережает мировую. Так, именно российские ученые предложили использовать одновременно несколько разных взаимоподдерживающих алгоритмов анализа последовательностей. Каждая из существующих программ по отдельности ошибается достаточно часто. Но если использовать несколько таких «слабых» программ одновременно, то там, где их предсказания совпадут, обнаружится истина. Например, человеческие гены удается неплохо предсказывать, если одновременно учитывать статистическое распределение нуклеотидов, сигналы сплайсинга и частоту использования кодонов.
Геномные исследования в России развиваются, несмотря на все трудности, с которыми сталкивается наука в нашей стране. И России они необходимы так же, как и другим странам.
Любая биологическая система стремится к поддержанию стабильности своего существования. Даже березовая роща контролирует внутренние условия – в ней и температура воздуха, и освещенность, и влажность воздуха отличаются от окружающих. Со всех видов естественный отбор собирает суровую дань, уничтожая носителей неблагоприятных мутаций, – это плата за приспособленность вида в целом.
К улучшению своей породы люди стремились во все времена. В древней Спарте сбрасывали «неудавшихся» младенцев со скалы. В 1930-х гг. в США с той же целью около 100 тыс. человек было подвергнуто принудительной стерилизации – с точки зрения генетики бессмысленной, т.к. подобные меры не снижают частоты проявления наследственных заболеваний в следующем поколении. В начале третьего тысячелетия человечество стремится взять под контроль собственные генетические процессы и вносить коррективы не ценой жизни носителя неблагоприятных мутаций, а подправляя их генетические тексты с учетом результатов исследований генома человека.

Таблица. Триплетный код (в скобках указана средняя частота встречаемости данного кодона на 1 тыс.) в геноме кишечной палочки
ТТТ (22) Фенилаланин |
ЦЦЦ (5) Пролин |
ААА (35) Лизин |
Человеку давно не давал покоя вопрос “Кто я?”, и в поисках ответа на него он на всякий случай торопился улучшить себя прикладной генетикой: древние греки убивали нездоровых детей, чтобы “очищать” популяцию. Выяснилось - абсолютно напрасно. Генетические дефекты внешне не видны, и какой-нибудь спартанский задохлик, если бы ему не свернули шею, мог бы наплодить олимпийских чемпионов. Перенесемся в год 2016. Вашему пышащему здоровьем годовалому младенцу составляют генетический паспорт, по которому видно, что у него есть 3 “больных” гена, свидетельствующие о предрасположенности к сердечно-сосудистым заболеваниям. Ему подбирается индивидуальная медикаментозная программа и диета, и он преспокойно доживает до 22 века благодаря открытиям, сделанным в рамках проекта “Геном человека”. Фантастика? Да нет, геномика.
Начало XXI века ознаменовалось серьезной биологической шумихой. В июне 2000 года на церемонии в Белом доме президент Клинтон торжественно замирил две соперничающие группы - компанию “Селера”, возглавляемую Крэгом Вентером, и межгосударственный проект HUGO под руководством директора национального Института генома Фрэнсиса Коллинза, и объявил, что после многих лет работы обе они, независимо друг от друга, вплотную подошли к расшифровке генома человека, то есть к составлению полной последовательности всех человеческих дезоксирибонуклеиновых кислот (ДНК).
Основной рабочей единицей любой живой системы является клетка. Вся информация, необходимая для жизнедеятельности клеток, хранится в ДНК. Геном - это полный набор ДНК организма. В человеческом геноме ДНК содержатся в 23 хромосомных парах. Хромосомы - это отдельные молекулы длиной от 50 млн до 250 млн базовых пар ДНК. Каждая хромосома несет в себе различные гены, которые представляют собой определенные последовательности базовых пар ДНК с закодированной информацией о порядке воспроизводства белков. Если информация не та - нормальные биологические функции будут нарушены, и возникнет болезнь.
В 1986 году американское министерство энергетики (а оно в США по указу Конгресса занимается широким разбросом технологий и, например, стояло у истоков создания ядерной бомбы) в партнерстве с Национальным Институтом здоровья объявило о запуске амбициозного проекта “Геном человека”, к участию в котором был приглашен ряд научных учреждений Японии, Англии, Франции, Германии и Китая. Целью проекта было создать базу данных, содержащую последовательность всех базовых пар ДНК и идентифицировать все человеческие гены. Попутно проект совершенствовал вычислительные механизмы для обеспечения будущих изысканий в биогенетике, определял коммерческие перспективы исследований и служил хорошей школой следующему поколению ученых-генетиков. В 1990 году Конгресс США начал финансировать работу проекта, рассчитанного на 15 лет.
Было немало скептиков, которые ставили под сомнение осуществление столь грандиозного проекта и его целесообразность. Первые несколько лет были потрачены на разработку методик и создание технологий, позволяющих подсчитывать и обрабатывать такое огромное количество данных. Поначалу идентификация одной базовой пары обходилась в $10, и специалист высокой квалификации мог просканировать до 10000 базовых пар в день. К моменту завершения проекта появились роботы, которые в состоянии обрабатывать 10000 пар в секунду, снизив стоимость идентификации до 5 центов.
Работа над проектом закипела намного энергичнее, когда в 1999 году биологический концерн “Селера Джиномикс” объявил о своем намерении самому вычленить все человеческие ДНК и начать продавать информацию о них фармацевтическим компаниям. Вооруженная обнародованными к тому времени наработками группы Коллинза, “Селера” собиралась провернуть всю работу за 3 года и утереть нос государственным бюрократам. Потенциальный позор вызвал усиленное вливание денег налогоплательщиков (проект к тому времени “кушал” по 1 млн долларов в день) и нелестную критику “Селеры”, рвущуюся к сверхприбылям, приторговывая достоянием всего человечества. Проект “Геном человека” заработал с такой скоростью, что черновой портрет человеческих ДНК был обнародован на 2 года раньше срока. Ученые из “Селеры” тут же отыскали в нем массу огрехов. Вмешательство администрации президента положило конец этим распрям, и в 2000 году Клинтон соединил руки Вентера и Коллинза сказав, что и государственный, и частный сектор разными путями пришли к похожим выводам, что успех второго был бы невозможен без наработок первого, и что благодаря участию “Селеры” результаты исследования быстрее преобразуются в конкретные плюсы для потребителей.
В апреле 2003 года проект, обошедшийся американской казне в 2,7 млрд долларов, был формально завершен на 2 года раньше срока. Президенты всех стран-участников проекта поздравили генетиков с победой. В распоряжение ученых всего мира поступил источник информации беспрецендентного объема, предопределяющий ход дальнейших изысканий на много лет вперед. Параллельно с человеческой были расшифрованы нуклеотидные последовательности многих других организмов, что дало толчок в развитии сравнительной генетики. Были быстро идентифицированы десятки тысяч генов, отвечающих за врожденные глухоту и слепоту, заболевания почек, рак груди, болезни костной ткани, инсульта, диабета и других заболеваний. Если раньше ученым потребовалось 9 лет работы, чтобы открыть ген, вызывающий фиброзно-кистозную дегенерацию, то, на основании опыта, полученного в ходе исследований генома, ген был найден всего за 9 дней.
Правда, когда все протрезвели от выпитого по случаю шампанского, выяснилось, что около 1% генома расшифровке не поддалось, что вероятность ошибок хоть и снизилась в 10 раз, но остановилась на уровне одной на 100.000 базовых пар ДНК и что пробелы в последовательности все-таки попадаются каждые 27 млн базовых пар. По-прежнему остался вопрос, сколько же генов делают человека человеком. В настоящее время предположительное количество колеблется на уровне 30 тысяч. Ожидается, что со временем оно вырастет до 40.000, но уж точно не до 100 тысяч, как предполагали многие. Таким образом, у человека лишь в 3 раза больше генов, чем у мухи дрозофилы, и лишь на пару сотен больше, чем у мыши.
Еще ученые узнали, что:
- В человеческом геноме 3 млрд нуклеотидных базовых пар.
- Наш ген состоит в среднем из 3 тысяч базовых пар; самый большой ген - дистрофин - состоит из 2,4 млн пар.
- Люди очень похожи - последовательность ДНК на 99,9% одинаковая у всех.
- Больше всего генов (2968) в самой большой человеческой хромосоме - 1, меньше всего (231) в самой маленькой хромосоме - Y.
- Около 2% генома содержит информацию по синтезу белков.
- Белки, а не гены, отвечают за выполнение жизненно-важных функций.
- В геноме насчитывается 30 тысяч генов и 400 тысяч белков.
Из последнего наблюдения делается вывод, что многообразие белков не объясняется результатами генетических исследований. Если геном остается сравнительно неизменяемым, то протеом находится в постоянном движении в зависимости от сигналов, поступающих как снаружи, так и изнутри клетки. Поведение каждого отдельно взятого белка определяется не только последовательностью генов, но и его взаимодействием с другими белками клетки. А про это нам известно совсем немного, и протеомика (изучение строения белка), похоже, будет орешек покрепче геномики (изучения строения генов).
Вооружившись шекспировской цитатой, “что в прошлом - лишь пролог”, руководители проекта заговорили о следующей, прикладной стадии исследований. Генетическое тестирование стало одним из первых коммерческих применений открытий в области генетики. Получив доступ к последовательности нуклеотидов, медики оказались в состоянии предсказывать предрасположенность к той или иной болезни и ее этапы, ставить правильный диагноз с максимально эффективным курсом лечения и минимальными побочными эффектами, разрабатывать новые лекарства на молекулярном уровне не столько для лечения, сколько для предотвращения заболеваний.
Естественно, что легче лечить болезни достаточно редкие, связанные с повреждением одного гена, а не многих. Но тут оказалось, что генотерапия в целом отстает от научных открытий, и далеко не все достижения могут быть успешно претворены в жизнь. Например, когда был выделен ген мышечной дистрофии Дюшена (смертельная болезнь, когда в клетках не вырабатывается дистрофин, без которого не сокращаются мышцы), стало очевидно, что нет эффективных способов доставки здорового гена во все клетки, и нет механизма, способного предотвратить его отторжение клетками.
Общество также оказалось неподготовленным к этическим и психологическим последствиям информационного взрыва. Когда стало известно, что в планы ученых входит исследование генов, определяющих этническую принадлежность, индивидуальные черты и форму поведения, присущие конкретному индивиду, и другая политически заряженная проблематика, возникли опасения, что генетическая информация может попасть в “чужие” руки, что страховые компании будут использовать ее в дискриминационных целях, и что люди не готовы узнать о себе всю генетическую правду. Завязался спор, насколько гены обуславливают наше поведение, и можем ли мы, изменяя их, изменить себя. Подняла новую голову старая гидра евгеники, проснулся генетический детерминизм.
Как часто бывает с важными открытиями, обнародование последовательности ДНК вызвало чуть ли не больше вопросов, чем дало ответов. Ведь если у нас всего в 3 раза больше генов, чем у фруктовой мушки, то что же тогда означает быть человеком? С трепетом будем ждать момента, когда раскодируют последовательность нуклеотидов шимпанзе, потому что вполне вероятно, что у людей с ними будет полное совпадение. А как же тогда гений Леонардо, Шопена и Достоевского? Неужели человек - это лишь сумма генов? Не слишком ли просто?
“Повода для беспокойства нет”, - в один голос говорят бывшие соперники Вентер и Коллинз, - “не следует впадать в крайности”. Геном человека был “человеческим” лишь 10 млн из 3 млрд лет эволюции. Поэтому 10% наших генов очень древние, и их мы делим с каждым живущим под солнцем живым организмом. Еще больше общих генов у нас с позвоночными, у млекопитающих с человеком еще больше сходства, а с шимпанзе мы почти идентичны. Если бы проект вычленял гены шимпанзе, то результаты бы получились почти такие же: к тому времени, когда мы слезли с деревьев, все наши гены уже были на своих местах.
То, что случилось с нами тогда, не было какой-то “генетической революцией”, потому что наш геном не несет в себе никаких ее следов. Во время палеолита сэволюционировал человеческий мозг и возникло человеческое сознание, без которого невозможны чисто человеческие замашки, вроде любви, духовности и способности помножить 2 на 2. Но если сознание тихо и без генетических сдвигов пришло к нам на такой поздней стадии эволюции, то что же вызвало его появление? Каков бы ни был этот механизм, а по поводу него ведется много споров, можно предположить, что наш разум развился в сложном взаимодействии генов с окружающей средой. Гены набросали план-схему, а наши родители, соплеменники и приобретаемый опыт дали ей плоть. Поэтому искать гены разума или интеллекта - это все равно, что искать второй концерт Рахманинова среди рояльных струн.
А в каком беспорядке природа содержит наш геном? Сравнивая наши хромосомы с хромосомами других живых существ, мы замечаем, что природа не только добавляла новые гены, но переставляла местами, удваивала, а то и просто выкидывала большие сегменты хромосом, проводя нечто вроде капитальной уборки каждый миллион лет. Дарвин в “Происхождении видов” писал о величии воплощения жизни. По результатам исследования человеческого генома, это воплощение носило довольно произвольный характер: природа по-своему понимала генетическое строительство, но кто первый бросит в нее камень, сказав, что конечный продукт не соответствует первоначальному замыслу?
К 50-летию открытия структуры ДНК
А.В. Зеленин
ГЕНОМ РАСТЕНИЙ
А. В. Зеленин
Зеленин Александр Владимирович
- д.б.н.,
заведующий лабораторией Института молекулярной биологии
им. В.А. Энгельгардта РАН.
Впечатляющие достижения программы "Геном человека", а также успехи работ по расшифровке так называемых сверхмалых (вирусы), малых (бактерии, дрожжи) и средних (круглый червь, дрозофила) геномов сделали возможным переход к широкомасштабному изучению крупных и сверхкрупных геномов растений . Насущная необходимость детального изучения геномов наиболее важных в хозяйственном отношении растений была подчеркнута на совещании по геномике растений, состоявшемся в 1997 г. в США [ , ]. За прошедшие с того времени годы достигнуты несомненные успехи в этой области. В 2000 г. появилась публикация о полном секвенировании (установление линейной последовательности нуклеотидов всей ядерной ДНК) генома горчицы малой - арабидопсиса, в 2001 г. - о предварительном (черновом) секвенировании генома риса. Неоднократно сообщалось о работах по секвенированию крупных и сверхкрупных геномов растений (кукуруза, рожь, пшеница), однако эти сообщения не содержали конкретной информации и носили, скорее, характер деклараций о намерении.
Предполагается, что расшифровка геномов растений откроет перед наукой и практикой широкие перспективы. Прежде всего выявление новых генов и цепочки их генетической регуляции позволит существенно повысить продуктивность растений за счет использования биотехнологических подходов. С обнаружением, выделением, размножением (клонированием) и секвенированием генов, отвечающих за такие важнейшие функции растительного организма, как размножение и продуктивность, процессы изменчивости, устойчивости к воздействию неблагоприятных факторов среды, а также гомологичное спаривание хромосом, связывают появление новых возможностей для усовершенствования селекционного процесса. Наконец, выделенные и клонированные гены можно использовать для получения трансгенных растений с принципиально новыми свойствами и анализа механизмов регуляции активности генов.
Важность изучения геномов растений подчеркивает и то обстоятельство, что до настоящего времени число локализованных, клонированных и секвенированных генов растений невелико и колеблется, по различным оценкам, между 800 и 1200. Это в 10-15 раз меньше, чем, например, у человека.
Несомненным лидером в широкомасштабном изучении геномов растений остаются США, хотя интенсивные исследования генома риса проводятся в Японии, а в последние годы и в Китае. В расшифровке генома арабидопсиса, кроме лабораторий США, приняли активное участие исследовательские группы Европы. Явное лидерство США вызывает серьезное беспокойство европейских ученых, которое они ясно выразили на совещании под многозначительным названием "Перспективы геномики в постгеномную эру", состоявшемся в конце 2000 г. во Франции. Опережение американской науки в изучении геномов сельскохозяйственных растений и создании трансгенных растительных форм, по мнению европейских ученых, грозит тем, что в не слишком отдаленном будущем (от двух до пяти десятилетий), когда рост численности населения поставит человечество перед лицом всеобщего продовольственного кризиса, европейская экономика и наука попадут в зависимость от американских технологий. В связи с этим объявлено о создании франко-германской научной программы по исследованию геномов растений ("Plantgene") и вложении в нее значительных средств.
Очевидно, что проблемы геномики растений должны привлечь пристальное внимание российских ученых и организаторов науки, а также руководящих инстанций, поскольку речь идет не только о научном престиже, но и о национальной безопасности страны. Через одно-два десятилетия продовольствие станет важнейшим стратегическим ресурсом.
ТРУДНОСТИ В ИЗУЧЕНИИ ГЕНОМОВ РАСТЕНИЙ
Изучение геномов растений - задача значительно более сложная, чем исследование генома человека и других животных . Это связано со следующими обстоятельствами:
огромными размерами геномов, достигающими для отдельных видов растений десятков и даже сотен миллиардов пар нуклеотидов (п.н.): геномы основных хозяйственно важных растений (кроме риса, льна и хлопка) по размерам либо близки к геному человека, либо превышают его во много раз (таблица);ХРОМОСОМНЫЕ ИССЛЕДОВАНИЯ ГЕНОМОВРезкими колебаниями числа хромосом у различных растений - от двух у некоторых видов до нескольких сотен у других, причем не удается выявить строгой корреляции между размером генома и числом хромосом;
Изобилием полиплоидных (содержащих более двух геномов на клетку) форм с близкими, но не идентичными геномами (аллополиплоидия);
Чрезвычайной обогащенностью геномов растений (до 99%) "незначащей" (некодирующей, то есть не содержащей генов) ДНК, что резко затрудняет стыковку (расположение в правильном порядке) отсеквенированных фрагментов в общий крупноразмерный участок ДНК (контиг);
Неполным (по сравнению с геномами дрозо-филы, человека и мыши) морфологическим, генетическим и физическим картированием хромосом;
Практической невозможностью выделять в чистом виде индивидуальные хромосомы с помощью методов, обычно применяемых с этой целью для хромосом человека и животных (сортировка в потоке и использование гибридов клеток);
Трудностью хромосомного картирования (определение расположения на хромосоме) отдельных генов с помощью гибридизации in situ , обусловленной как высоким содержанием в геномах растений "незначащей" ДНК, так и особенностями структурной организации хромосом растений;
Эволюционной отдаленностью растений от животных, что серьезно осложняет использование для изучения геномов растений сведений, полученных при секвенировании генома человека и других животных;
Длительным процессом размножения большинства растений, что существенно замедляет их генетический анализ.
Хромосомные (цитогенетические) исследования геномов вообще и растений в частности имеют длинную историю. Термин "геном" был предложен для обозначения гаплоидного (единичного) набора хромосом с содержащимися в них генами в первой четверти XX в., то есть задолго до установления роли ДНК как носителя генетической информации .
Описание генома нового, ранее генетически не изученного многоклеточного организма обычно начинают с исследования и описания полного набора его хромосом (кариотипа). Это, разумеется, относится и к растениям, огромное множество которых еще даже не начали изучать.
Уже на заре хромосомных исследований проводили сравнение геномов родственных видов растений на основе анализа мейотической конъюгации (объединения гомологичных хромосом) у межвидовых гибридов. За прошедшие 100 лет возможности хромосомного анализа резко расширились. Сейчас для характеристики геномов растений используют более совершенные технологии: различные варианты так называемого дифференциального окрашивания, позволяющего по морфологическим признакам идентифицировать индивидуальные хромосомы; гибридизацию in situ, дающую возможность локализовать конкретные гены на хромосомах; биохимические исследования клеточных белков (электрофорез и иммунохимия) и, наконец, комплекс методов, основанных на анализе хромосомной ДНК вплоть до ее секвенирования.

Рис. 1. Кариотипы хлебных злаков а - рожь (14 хромосом), б - твердая пшеница (28 хромосом), в - мягкая пшеница (42 хромосомы), г - ячмень (14 хромосом)В течение многих лет изучаются кариотипы хлебных злаков, прежде всего пшеницы и ржи. Интересно, что у разных видов этих растений число хромосом различно, но всегда кратно семи. Отдельные виды хлебных злаков можно надежно распознать по их кариотипу. Например, геном ржи состоит из семи пар крупных хромосом, имеющих на своих концах интенсивно окрашенные гетерохроматические блоки, часто называемые сегментами, или бэндами (рис. 1, а). Геномы пшеницы насчитывают уже 14 и 21 пару хромосом (рис. 1, б, в), причем распределение в них гетерохроматических блоков не такое, как в хромосомах ржи. Различаются между собой и отдельные геномы пшеницы, получившие обозначение А, В и D. Возрастание числа хромосом с 14 до 21 приводит к резкому изменению свойств пшеницы, что нашло отражение в их названиях: твердая, или макаронная, пшеница и мягкая, или хлебная, пшеница. За приобретение мягкой пшеницей высоких хлебопекарных свойств ответственен геном D, содержащий гены белков клейковины, которая придает тесту так называемую всхожесть. Именно этому геному уделяется особое внимание при селекционном улучшении хлебных пшениц. Другой 14-хромосомный злак - ячмень (рис. 1, г) обычно не используют для приготовления хлеба, однако он служит основным сырьем для изготовления таких распространенных продуктов, как пиво и виски.
Интенсивно изучаются хромосомы некоторых дикорастущих растений, используемых для улучшения качества важнейших сельскохозяйственных видов, например диких сородичей пшеницы - эгилопсов . Новые растительные формы создаются путем скрещивания (рис. 2) и отбора. В последние годы значительное усовершенствование методики исследований позволило приступить к изучению геномов растений, особенности кариотипов которых (главным образом мелкие размеры хромосом) делали их ранее недоступными для хромосомного анализа. Так, лишь недавно были впервые идентифицированы все хромосомы хлопка, ромашки и льна .

Рис. 2. Кариотипы пшеницы и гибрида пшеницы с эгилопсом
а - гексаплоидная мягкая пшеница (Triticum astivum ), состоящая из А, В и О геномов; б - тетраплоидная пшеница (Triticum timopheevi ), состоящая из А и G геномов. содержит гены устойчивости к большинству болезней пшеницы; в - гибриды Triticum astivum х Triticum timopheevi , устойчивые к мучнистой росе и ржавчине, отчетливо видно замещение части хромосомПЕРВИЧНАЯ СТРУКТУРА ДНК
По мере развития молекулярной генетики расширилось само понятие генома. Сейчас этот термин трактуется как в классическом хромосомном, так и в осовремененном молекулярном смысле: весь генетический материал отдельного вируса, клетки и организма . Естественно, что вслед за изучением полной первичной структуры геномов (так часто называют полную линейную последовательность оснований нуклеиновых кислот) ряда микроорганизмов и человека на очередь встал вопрос о секвенировании геномов растений.
Из множества растительных организмов для исследования были выбраны два - арабидопсис, представляющий класс двудольных (размер генома 125 млн. п.н.), и рис из класса однодольных (420-470 млн. п.н.). Эти геномы невелики по сравнению с геномами других растений и содержат сравнительно немного повторяющихся участков ДНК. Такие особенности давали надежду на то, что выбранные геномы окажутся доступными для относительно быстрого определения их первичной структуры.

Рис. 3. Арабидопсис - горчица малая - мелкое растение из семейства крестоцветных (Brassicaceae ). На пространстве, равном по площади одной странице нашего журнала, можно вырастить до тысячи индивидуальных организмов арабидопсисаОснованием для выбора арабидопсиса послужили не только небольшие размеры его генома, но и мелкие размеры организма, что позволяет легко выращивать его в лабораторных условиях (рис. 3). Принимали во внимание его короткий репродуктивный цикл, благодаря чему можно быстро проводить опыты по скрещиванию и отбору, детально изученную генетику, легкость осуществления манипуляций со сменой условий произрастания (изменение солевого состава почвы, добавление разных питательных веществ и т.д.) и с испытанием действия на растения различных мутагенных факторов и патогенов (вирусы, бактерии, грибы). Арабидопсис не имеет хозяйственной ценности, поэтому его геном, наряду с геномом мыши, получил название справочного, или, что менее точно, модельного.*
* Появление в отечественной литературе термина "модельный геном" - результат неточного перевода английского словосочетания model genome. Слово "model" означает не только прилагательное "модельный", но и существительное "образец", "эталон", "модель". Правильнее было бы говорить о геноме-образце, или справочном геноме.Интенсивная работа по секвенированию генома арабидопсиса была начата в 1996 г. международным консорциумом, в который вошли научные учреждения и исследовательские группы из США, Японии, Бельгии, Италии, Великобритании и Германии. В декабре 2000 г. стала доступной обширная информация, подводившая итоги определения первичной структуры генома арабидопсиса . Для секвенирования использовали классическую, или иерархическую, технологию: сначала изучали отдельные небольшие участки генома, из которых составляли более крупные участки (контиги), а на финальном этапе - структуру индивидуальных хромосом. Ядерная ДНК генома арабидопсиса распределена между пятью хромосомами. В 1999 г. были опубликованы результаты секвенирования двух хромосом, а появление в печати сведений о первичной структуре остальных трех завершило секвенирование всего генома.
Из 125 млн. пар нуклеотидов определена первичная структура 119 млн., что составляет 92% всего генома. Лишь 8% генома арабидопсиса, содержащих крупные блоки повторяющихся участков ДНК, оказались недоступными для изучения. По полноте и тщательности секвенирования геномов эукариот арабидопсис остается пока в первой тройке чемпионов наряду с одноклеточным дрожжевым организмом Saccharomyces cerevisiae и многоклеточным организмом животного Саеnorhabditis elegance (см. табл.).
В геноме арабидопсиса обнаружено около 15 тыс. индивидуальных генов, кодирующих белки. Приблизительно 12 тыс. из них содержатся в виде двух копий на гаплоидный (единичный) геном, так что общее число генов составляет 27 тыс. Число генов у арабидопсиса не сильно отличается от числа генов у таких организмов, как человек и мышь, однако размеры его генома в 25-30 раз меньше. С этим обстоятельством связаны важные особенности в структуре отдельных генов арабидопсиса и общей структуры его генома.
Гены арабидопсиса компактны, содержат лишь несколько экзонов (участков, кодирующих белки), разделенных короткими (около 250п.н.) некодирующими отрезками ДНК (интронами). Промежутки между отдельными генами составляют в среднем 4.6 тыс. пар нуклеотидов. Для сравнения укажем, что гены человека содержат многие десятки и даже сотни экзонов и интронов, а межгенные участки имеют размеры от 10 тыс. пар нуклеотидов и более. Предполагают, что наличие небольшого компактного генома способствовало эволюционной устойчивости арабидопсиса, поскольку его ДНК в меньшей степени становилась мишенью для воздействия различных повреждающих агентов, в частности, для внедрения в геном вирусоподобных повторяющихся фрагментов ДНК (транспозонов).
Из других молекулярных особенностей генома арабидопсиса следует отметить обогащенность экзонов гуанином и цитозином (44% в экзонах и 32% в интронах) по сравнению с генами животных, а также присутствие дважды повторенных (дуплицированных) генов. Предполагают, что такое удвоение произошло в результате четырех одномоментных событий, заключавшихся в удвоении (повторении) части генов арабидопсиса, или слияния родственных геномов. Эти события, имевшие место 100-200 млн. лет назад, - проявление общей тенденции к полиплоидизации (кратному увеличению числа геномов в организме), характерной для геномов растений. Однако некоторые факты показывают, что у арабидопсиса удвоеннные гены неидентичны и функционируют по-разному, что может быть связано с мутациями в их регуляторных участках.
Еще одним объектом полного секвенирования ДНК стал рис . Геном этого растения тоже невелик (12 хромосом, дающих в сумме 420-470 млн. п.н.), всего в 3.5 раза больше, чем у арабидопсиса. Однако, в отличие от арабидопсиса, рис имеет огромное хозяйственное значение, являясь основой питания для более чем половины человечества, поэтому в улучшении его свойств кровно заинтересованы не только миллиарды потребителей, но и многомиллионная армия людей, активно вовлеченная в весьма трудоемкий процесс его выращивания.
Отдельные исследователи приступили к изучению генома риса еще в 80-х годах прошлого столетия, но серьезного масштаба эти работы достигли лишь в 90-х. В 1991 г. в Японии была создана программа по расшифровке структуры генома риса, объединившая усилия многих исследовательских групп. В 1997 г. на базе этой программы был организован Международный проект "Геном риса" . Его участники решили сконцентрировать усилия на секвенировании одного из подвидов риса (Oriza sativajaponica ), в изучении которого к тому времени уже были достигнуты значительные успехи. Серьезным стимулом и, образно выражаясь, путеводной звездой для такой работы стала программа "Геном человека" .
В рамках этой программы прошла апробацию стратегия "похромосомного" иерархического разделения генома, которую участники международного консорциума использовали при расшифровке генома риса. Однако, если при изучении генома человека с помощью различных приемов выделяли фракции отдельных хромосом, то материал, специфичный для индивидуальных хромосом риса и их отдельных участков, получали методом лазерной микродиссекции (вырезания микроскопических объектов). На предметном стекле микроскопа, где находятся хромосомы риса, под воздействием лазерного луча выжигается все, кроме хромосомы или ее участков, намеченных для анализа. Оставшийся материал используют для клонирования и секвенирования.
Опубликованы многочисленные сообщения о результатах секвенирования отдельных фрагментов генома риса, осуществленного с высокой точностью и детальностью, характерной для иерархической технологии. Считали, что определение полной первичной структуры генома риса будет завершено к концу 2003-середине 2004 г. и результаты вместе с данными по первичной структуре генома арабидопсиса будут широко использоваться в сравнительной геномике других растений.
Однако в начале 2002 г. две исследовательские группы - одна из Китая, другая из Швейцарии и США - опубликовали результаты полного чернового (приблизительного) секвенирования генома риса, выполненного с помощью технологии тотального клонирования . В отличие от поэтапного (иерархического) изучения, тотальный подход основан на одномоментном клонировании всей геномной ДНК в одном из вирусных или бактериальных векторов и получении значительного (огромного для средних и крупных геномов) количества отдельных клонов, содержащих различные отрезки ДНК. На основании анализа этих секвенированных участков и наложения друг на друга идентичных концевых участков ДНК образуется контиг - цепочка стыкованных между собой последовательностей ДНК. Общий (суммарный) контиг представляет собой первичную структуру всего генома или, по крайней мере, индивидуальной хромосомы.
В таком схематичном изложении стратегия тотального клонирования кажется несложной. На деле она встречает серьезные трудности, связанные с необходимостью получения огромного количества клонов (принято считать, что изучаемый геном или его участок должен быть перекрыт клонами, по крайней мере, 10 раз), гигантским объемом секвенирования и чрезвычайно сложной работой по стыковке клонов, требующей участия специалистов по биоинформатике. Серьезным препятствием на пути тотального клонирования служат разнообразные повторяющиеся участки ДНК, число которых, как уже упоминалось, резко возрастает по мере увеличения размера генома. Поэтому стратегию тотального секвенирования используют преимущественно при изучении геномов вирусов и микроорганизмов, хотя она и была успешно применена для исследования генома многоклеточного организма -дрозофилы.
Результаты тотального секвенирования этого генома были "наложены" на огромный массив сведений о его хромосомной, генной и молекулярной структуре, полученных за почти 100-летний период изучения дрозофилы. И все же по степени секвенированности геном дрозофилы (66% общего размера генома) значительно уступает геному арабидопсиса (92%), несмотря на достаточно близкие их размеры - 180 млн. и 125 млн. пар нуклеотидов соответственно. Поэтому недавно предложено называть смешанной технологию, с помощью которой проводилось секвенирование генома дрозофилы.
Для секвенирования генома риса упомянутые выше исследовательские группы взяли два его подвида, наиболее широко культивируемые в азиатских странах, - Oriza saliva L. ssp indicaj и Oriza saliva L. sspjaponica. Результаты их исследований во многом совпадают, но во многом и различаются. Так, представители обеих групп заявили, что ими достигнуто перекрывание контигами приблизительно 92-93% генома. Показано, что около 42% генома риса представлено короткими повторами ДНК, состоящими из 20 пар нуклеотидов, и большинство подвижных ДНК-элементов (транспозонов) находится в межгенных участках. Однако сведения о размерах генома риса существенно различаются.
Для японского подвида размер генома определен равным 466 млн. пар нуклеотидов, а для индийского - 420 млн. Причина такого расхождения не ясна. Оно может быть следствием различных методических подходов в определении размеров некодирующей части геномов, то есть не отражать истинного положения дел. Но не исключено, что 15%-ное различие в размере изученных геномов действительно существует.
Второе серьезное расхождение выявилось в числе обнаруженных генов: для японского подвида - от 46022 до 55615 генов на геном, а для индийского - от 32000 до 50000. Причина такого расхождения не ясна.
Неполнота и противоречивость полученных сведений отмечена в комментариях к опубликованным статьям . Здесь же высказана надежда, что пробелы в знаниях генома риса будут устранены при сопоставлении данных "чернового секвенирования" с результатами детального, иерархического секвенирования, проводимого участниками Международного проекта "Геном риса".
СРАВНИТЕЛЬНАЯ И ФУНКЦИОНАЛЬНАЯ ГЕНОМИКА РАСТЕНИЙ
Полученные обширные данные, половина из которых (результаты китайской группы) общедоступны, несомненно, открывают широкие перспективы как для изучения генома риса, так и для геномики растений в целом. Сравнение свойств геномов арабидопсиса и риса показало, что большая часть генов (до 80%), выявленных в геноме арабидопсиса, обнаружена и в геноме риса, однако приблизительно для половины генов, обнаруженных у риса, пока не удалось найти аналогов (ортологов) в геноме арабидопсиса. В то же время 98% генов, первичная структура которых установлена для других злаков, выявлены в геноме риса.
Вызывает недоумение существенное (почти в два раза) расхождение в числе генов у риса и арабидопсиса. При этом данные черновой расшифровки генома риса, полученные с помощью тотального секвенирования, практически не сопоставлены с обширными результатами изучения генома риса методом иерархического клонирования и секвенирования, то есть не осуществлено то, что сделано в отношении генома дрозофилы. Поэтому остается неясным, отражает ли различие числа генов у арабидопсиса и риса истинное положение дел или же оно объясняется различием в методических подходах.
В отличие от генома арабидопсиса, сведения о генах-двойниках в геноме риса не приведены. Не исключено, что их относительное количество может быть больше у риса, чем у арабидопсиса. В пользу такой возможности косвенно свидетельствуют данные о наличии полиплоидных форм риса. Большей ясности в этом вопросе можно ожидать после завершения Международного проекта "Геном риса" и получения детальной картины первичной структуры ДНК этого генома. Серьезные основания для такой надежды дает тот факт, что после выхода в свет работ о черновом секвенировании генома риса резко увеличилось число публикаций о структуре этого генома, в частности, появились сведения о детальном секвенировании его 1 и 4 хромосом.
Знание, хотя бы приблизительное, числа генов у растений имеет принципиальное значение для сравнительной геномики растений. Вначале считали, что поскольку по своим фенотипическим признакам все цветковые растения весьма близки друг к другу, так же близки должны быть и их геномы. И если мы изучим геном арабидопсиса, то получим сведения и о большинстве геномов других растений. Косвенным подтверждением такого допущения служат результаты секвенирования генома мыши , который удивительно близок к геному человека (около 30 тыс. генов, из которых различными оказалась лишь 1 тыс.).
Можно предположить, что причина различий геномов арабидопсиса и риса кроется в их принадлежности к разным классам растений - двудольным и однодольным. Чтобы прояснить этот вопрос, крайне желательно знать хотя бы черновую первичную структуру еще какого-нибудь однодольного растения. Наиболее реальным кандидатом может стать кукуруза, геном которой приблизительно равен геному человека, но все-таки значительно меньше геномов других злаков. Продовольственное значение кукурузы общеизвестно.
Огромный материал, полученный в результате секвенирования геномов арабидопсиса и риса, постепенно становится основой для широкомасштабного изучения геномов растений методами сравнительной геномики. Такие исследования имеют общебиологическое значение, так как позволяют установить главные принципы организации генома растений в целом и их отдельных хромосом, выявить общие черты структуры генов и их регуляторных участков, рассмотреть соотношение функционально активной (генной) части хромосомы и различных не кодирующих белки межгенных участков ДНК. Сравнительная генетика приобретает все большее значение и для развития функциональной геномики человека. Именно для проведения сравнительных исследований осуществлено секвенирование геномов рыбы фугу и мыши.
Не менее важно изучение отдельных генов, ответственных за синтез индивидуальных белков, определяющих конкретные функции организма. Именно в обнаружении, выделении, секвенировании и установлении функции отдельных генов состоит практическое, в первую очередь медицинское, значение программы "Геном человека". Это обстоятельство несколько лет назад отметил Дж. Уотсон, подчеркнувший, что программа "Геном человека" будет завершена лишь тогда, когда определят функции всех генов человека.
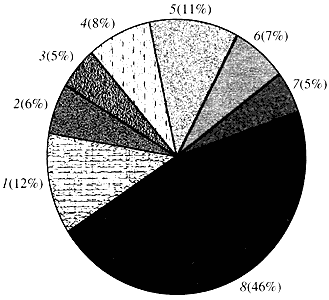
Рис. 4. Классификация по функции генов арабидопсиса
1 - гены роста, деления и синтеза ДНК; 2 - гены синтеза РНК (транскрипция); 3 - гены синтеза и модификации белков; 4 - гены развития, старения и смерти клеток; 5 - гены клеточного метаболизма и энергетического обмена; 6 - гены межклеточного взаимодействия и передачи сигнала; 7 - гены обеспечения прочих клеточных процессов; 8 - гены с неизвестной функциейЧто касается функции генов растений, то мы знаем о них менее одной десятой того, что нам известно о генах человека. Даже у арабидопсиса, геном которого по степени изученности намного превосходит геном человека, функция почти половины его генов остается неизвестной (рис. 4). Между тем у растений, кроме генов, общих с животными, имеется значительное число генов, специфичных только (или, по крайней мере, преимущественно) для них. Речь идет о генах, вовлеченных в транспорт воды и синтез клеточной стенки, отсутствующей у животных, о генах, обеспечивающих образование и функционирование хлоропластов, фотосинтез, фиксацию азота и синтез многочисленных ароматических продуктов. Этот перечень можно продолжить, но уже сейчас ясно, сколь сложная задача стоит перед функциональной геномикой растений.
Полное секвенирование генома дает близкие к истинным сведения об общем количестве генов данного организма, позволяет поместить в банки данных более или менее подробные и достоверные сведения об их структуре, облегчает работу по выделению и изучению индивидуальных генов. Однако секвенирование генома отнюдь не означает установления функции всех генов.
Один из наиболее перспективных подходов функциональной геномики базируется на выявлении работающих генов, на которых идет транскрипция (считывание) мРНК. Этот подход, в том числе использующий современную технологию микрочипов, позволяет одновременно выявлять до десятков тысяч функционирующих генов. Недавно с помощью такого подхода начато изучение геномов растений. Для арабидопсиса удалось получить около 26 тыс. индивидуальных транскриптов, что резко облегчает возможность определения функции практически всех его генов . У картофеля удалось выявить около 20000 тыс. работающих генов, важных для понимания как процессов роста и формирования клубня, так и процессов заболевания картофеля. Предполагается, что это знание позволит повысить устойчивость одного из важнейших пищевых продуктов к возбудителям заболеваний.
Логичным развитием функциональной геномики стала протеомика. Эта новая область науки изучает протеом, под которым обычно подразумевают полный набор белков в клетке в конкретный момент. Такой набор белков, отражающий функциональное состояние генома, все время меняется, тогда как геном остается неизменным.
Изучение белков уже давно используют для суждений об активности геномов растений. Как известно, ферменты, имеющиеся у всех растений, различаются у отдельных видов и сортов последовательностью аминокислот. Такие ферменты, с одинаковой функцией, но разной последовательностью отдельных аминокислот, называют изоферментами. У них разные физико-химические и иммунологические свойства (молекулярная масса, заряд), что можно выявить с помощью хро-матографии или электрофореза. В течение многих лет эти методы успешно использовали для изучения так называемого генетического полиморфизма, то есть различий между организмами, сортами, популяциями, видами, в частности пшеницы и родственных форм злаков. Однако в последнее время в связи с быстрым развитием методов анализа ДНК, включая секвенирование, изучение белкового полиморфизма оказалось замененным исследованием полиморфизма ДНК. Впрочем, прямое изучение спектров запасных белков (проламины, глиадины и др.), определяющих основные питательные свойства злаков, остается важным и надежным способом генетического анализа, селекции и семеноводства сельскохозяйственных растений.
Знание генов, механизмов их экспрессии и регуляции чрезвычайно важно для развития биотехнологии и получения трансгенных растений. Известно, что впечатляющие успехи в этой области вызывают неоднозначную реакцию экологической и медицинской общественности. Однако есть область биотехнологии растений, где эти страхи, если не совсем беспочвенны, то, во всяком случае, представляются малосущественными. Речь идет о создании трансгенных технических растений, не используемых в качестве пищевых продуктов. Недавно в Индии собран первый урожай трансгенного хлопка, устойчивого к ряду заболеваний . Имеются сведения о введении в геном хлопка специальных генов, кодирующих пигментные белки, и получении волокон хлопка, не нуждающихся в искусственном окрашивании. Другая техническая культура, которая может оказаться объектом эффективной генной инженерии, - это лен. Его использование как альтернативы хлопку для получения текстильного сырья обсуждается в последнее время . Эта проблема чрезвычайно важна для нашей страны, утратившей собственные источники хлопкового сырья.
ПЕРСПЕКТИВЫ ИЗУЧЕНИЯ ГЕНОМОВ РАСТЕНИЙ
Очевидно, что структурные исследования геномов растений будут базироваться на подходах и методах сравнительной геномики с использованием в качестве основного материала результатов расшифровки геномов арабидопсиса и риса. Существенную роль в развитии сравнительной геномики растений будут, без сомнения, играть сведения, которые рано или поздно предоставит тотальное (черновое) секвенирование геномов других растений. При этом сравнительная геномика растений будет основываться на установлении генетических взаимосвязей отдельных локусов и хромосом, относящихся к разным геномам. Речь пойдет не столько об общей геномике растений, сколько об избирательной геномике отдельных хромосомных локусов. Так, недавно было показано, что ген, ответственный за яровизацию, расположен в локусе VRn-AI хромосомы 5А гексаплоидной пшеницы и локусе Hd-6 хромосомы 3 риса.
Развитие этих исследований явится мощным толчком к идентификации, выделению и секвени-рованию многих функционально важных генов растений, в частности генов, ответственных за устойчивость к болезням, засухоустойчивость, приспособленность к различным условиям произрастания. Все шире будет использоваться функциональная геномика, основанная на массовом выявлении (скринировании) генов, функционирующих в растениях.
Можно предвидеть дальнейшее совершенствование хромосомных технологий, прежде всего метода микродиссекции. Его использование резко расширяет возможности геномных исследований, не требуя огромных затрат, как, например, тотальное секвенирование геномов. Получит дальнейшее распространение метод локализации на хромосомах растений отдельных генов с помощью гибридизации in situ. В настоящий момент его применение ограничено огромным числом повторяющихся последовательностей в геноме растений, а возможно, и особенностями структурной организации хромосом растений.
Хромосомные технологии в обозримом будущем приобретут большое значение и для эволюционной геномики растений. Эти технологии, относительно недорогие, позволяют быстро оценивать внутри- и межвидовую вариабельность, изучать сложные аллополиплоидные геномы тетраплоидной и гексаплоидной пшеницы, тритикале; анализировать эволюционные процессы на хромосомном уровне; исследовать образование синтетических геномов и введение (интрогрессия) чужеродного генетического материала; выявлять генетические взаимоотношения между индивидуальными хромосомами различных видов.
Изучение кариотипа растений с помощью классических цитогенетических методов, обогащаемых молекулярно-биологическим анализом и компьютерными технологиями, будет использоваться для характеристики генома. Это особенно важно для изучения стабильности и изменчивости кариотипа на уровне не только отдельных организмов, но и популяции, сорта и вида. Наконец, трудно представить, каким образом можно оценить число и спектры хромосомных перестроек (аберрации, мосты) без применения методов дифференциального окрашивания. Такие исследования крайне перспективны для мониторинга окружающей среды по состоянию генома растений.
В современной России вряд ли будет проводиться прямое секвенирование геномов растений. Такие работы, требующие крупных вложений, непосильны для нашей нынешней экономики. Между тем сведений о строении геномов арабидопсиса и риса, полученных мировой наукой и доступных в международных банках данных, достаточно для развития отечественной геномики растений. Можно предвидеть расширение исследований геномов растений, основанных на подходах сравнительной геномики, для решения конкретных задач селекции и растениеводства, а также изучения происхождения различных видов растений, имеющих важное хозяйственное значение.
Можно полагать, что в отечественной селекционной практике и растениеводстве будут широко использоваться такие геномные подходы, как генетическое типирование (RELF, RAPD, AFLP-анализы и т.п.), вполне доступные для нашего бюджета. Параллельно с прямыми методами определения ДНК-полиморфизма для решения проблем генетики и селекции растений будут применяться подходы, основанные на изучении белкового полиморфизма, в первую очередь запасных белков злаков. Широкое применение получат хромосомные технологии. Они относительно недороги, их развитие требует вполне умеренных вложений. В области хромосомных исследований отечественная наука не уступает мировой.
Следует подчеркнуть, что наша наука внесла заметный вклад в становление и развитие геномики растений [ , ].
Основополагающую роль сыграл Н.И. Вавилов (1887-1943).
В молекулярной биологии и геномике растений очевиден пионерский вклад А.Н. Белозерского (1905-1972).
В области хромосомных исследований необходимо отметить работы выдающегося генетика С.Г. Навашина (1857-1930), впервые обнаружившего у растений спутниковые хромосомы и доказавшего, что можно различать отдельные хромосомы по особенностям их морфологии.
Другой классик российской науки Г.А. Левицкий (1878-1942) детально описал хромосомы ржи, пшеницы, ячменя, гороха и сахарной свеклы, ввел в науку термин "кариотип" и развил учение о нем.
Современные специалисты, опираясь на достижения мировой науки, могут внести заметный вклад в дальнейшее развитие генетики и геномики растений.
Автор выражает сердечную благодарность академику Ю.П. Алтухову за критическое обсуждение статьи и ценные советы.Работа коллектива, возглавляемого автором статьи, выполнена при поддержке Российского фонда фундаментальных исследований (гранты № 99-04-48832; 00-04-49036; 00-04-81086), Программы Президента Российской Федерации по поддержке научных школ (гранты № 00-115-97833 и НШ-1794.2003.4) и Программы Российской академии наук "Молекулярно-генетические и хромосомные маркеры в разработке современных методов селекции и семеноводства".
ЛИТЕРАТУРА
1. Зеленин А.В., Бадаева Е.Д., Муравенко О.В.
Введение
в геномику растений // Молекулярная биология. 2001. Т. 35. С. 339-348. 2. Pen E.
Bonanza for Plant Genomics // Science.
1998. V. 282. P. 652-654. 3. Plant genomics // Proc. Natl. Acad. Sci. USA. 1998.
V. 95. P. 1962-2032. 4. Картель Н.А. и др.
Генетика. Энциклопедический
словарь. Минск: Technologia, 1999. 5. Badaeva E.D., Friebe B., Gill B.S.
1996. Genome
differentiation in Aegilops. 1. Distribution of highly repetitive DNA sequences
on chromosomes of diploid species // Genome. 1996. V. 39. P. 293-306. История
хромосомного анализа // Биол. мембраны. 2001. Т. 18. С. 164-172.
Ученые из США создали алгоритм, способный предсказывать по геному, как в живом организме работают транскрипционные факторы - белки, управляющие синтезом других белков. Исследование опубликовано в PNAS.
Хотя последовательности ДНК многих организмов уже прочитаны, ученые еще не до конца понимают, как они работают. Одной из таких загадок долгое время оставались Hox-гены, которые управляют ростом организма и развитием его частей в правильных местах. Именно они «приказывают» голове у личинки дрозофилы расти спереди, определяют, где и в каком количестве появятся крылья и ноги. Есть эта группа генов и у млекопитающих.
Hox-гены кодируют не простые белки, а особенные, которые называются транскрипционными факторами. Эти белки работают «выключателями» для многих других генов, присоединяясь к специальным участкам ДНК, чтобы усилить или запретить считывание последовательности. Это и позволяет Hox-генам «дирижировать» развитием эмбриона. Однако ученые заметили странную особенность: хотя каждый Hox-ген отвечает за рост и развитие разных частей организма, все они сильно связываются с одними и теми же последовательностями ДНК.
В 2015 году генетики из Колумбийского университета обнаружили, что эти транскрипционные факторы связываются и со множеством других участков ДНК, но намного слабее. Ученые поняли, что эти слабые связывания – ключ к пониманию работы Hox-генов. Однако найти их все в геноме было непросто. Для этого генетики создали новый метод секвенирования (прочтения последовательностей ДНК), который назвали SELEX-seq. Для этого подхода нужно было много раз подряд секвенировать один и тот же участок, однако он не давал информации о важных участках со слабым связыванием. «Это было словно прогонять один и тот же абзац через Google-переводчик снова и снова, но в конце получать только 10% слов, переведенных аккуратно», – прокомментировал работу один из авторов технологии, Хиггинсовский профессор биохимии и молекулярной биофизики в Колумбийском университете Ричард Манн.
Исследователи создали новый алгоритм, позволяющий понять, почему ДНК-последовательности вели себя в этом эксперименте именно так. Алгоритм назвали No Read Left Behind, или NRLB (буквально «ни одно прочтение не забыто»). Этот алгоритм стал первым количественным методом, способным оценить силу связывания участков ДНК с транскрипционными факторами. Кроме того, он смог точно предсказать влияние определенных мутаций на уровень экспрессии генов в эмбрионах дрозофилы даже для слабо связывающихся участков.
Транскрипционных факторов в геноме около 10%, и их сила связывания с разными последовательностями может варьироваться в тысячи раз. Поэтому работа важна не только в контексте изучения Hox-генов, но и для нашего понимания того, как функционирует геном.
Понравился материал? в «Мои источники» Яндекс.Новостей и читайте нас чаще.
В научных кругах несколько лет назад ходила байка, что Господь, создавая человека, действовал как самый настоящий программист. Программа, как правило, занимает гораздо меньше объема, чем комментарии к ней. В геноме оказалось буквально то же самое: только спустя десятилетие после прочтения человеческой ДНК «по буквам» ученые начали разбираться в «комментариях» к ней. Они нашли в геноме миллионы регуляторов работы генов и даже, судя по полученным результатам, выяснили, как они действуют. Таким образом, удалось получить массу информации о генетических заболеваниях.
В начале сентября в научном мире случилась сенсация. Практически одновременно были опубликованы 30 с лишним статей с результатами одного из проектов под названием ENCODE. Из них шесть статей появились в Nature, две — в Science и 24 - в журналах Genome Re search и Genome Biology.
Подобного всплеска молекулярно-генетических публикаций не случалось уже давно. Cуть проекта ENCODE состоит в том, что ученые исследовали ту часть ДНК, которую часто называют «мусорной» (junk DNA). Почему «мусорной»? Дело в том, что она не содержит генов, а значит, не кодирует белков. И было совершенно непонятно, для чего она нужна.
Но «мусор» оказался поистине золотым. Теперь, как говорит доктор Юан Бирни (Ewan Birney) из Европейского института биоинформатики в Хьюстоне (Великобритания), координатор анализа данных по проекту, термин «мусорная ДНК» пора выбросить в мусорную корзину.
Строение ДНК
ДНК (дезоксирибонуклеиновая кислота) - длинная полимерная молекула, состоящая из повторяющихся блоков, нуклеотидов. Каждый нуклеотид состоит из азотистого основания, сахара (дезоксирибозы) и фосфатной группы. Сама молекула имеет форму спирали из двух цепей, ориентированных азотистыми основаниями друг к другу. В ДНК четыре вида азотистых оснований (аденин, гуанин, тимин и цитозин), они соединяются попарно друг с другом водородными связями по принципу комплементарности (аденин соединяется с тимином, гуанин с цитозином).
Прочитан не значит расшифрован
В 2000 году мир узнал о том, что ученые прочитали геном человека. Точнее, в тот момент они всего лишь получили «черновик», а о полном прочтении объявили в 2003 году. В СМИ для этой работы использовали фразу «расшифровка генома», хотя она не совсем корректно описывает результат многолетней работы международного консорциума.
То, что получили, представляет собой не расшифровку, а самую настоящую шифровку. Исследователи секвенировали человеческую ДНК, то есть распознали последовательность из 3 млрд букв (А, С, T, G), обозначающих составные «кирпичики» ДНК - нуклеотиды. Двойная спираль молекулы, в которой закодирована наследственная информация человека, превратилась в длиннейшую телетайпную ленту с буквами, и некоторые из них складывались в «слова» - гены.
Но «слов-то» оказалось относительно немного! Общее количество генов в геноме человека ученые оценивают всего в 20-25 тыс. Эта цифра совсем не поражает воображение, если сравнивать, например, с мухой дрозофилой, у которой 14 тыс. генов. А у крошечного пресноводного рачка дафнии насчитали более 30 тыс. генов - на сегодня он остается чемпионом в этой номинации. Но мы-то посложнее устроены, чем дафния, один мозг человеческий чего стоит. Стало ясно, что особенность генетического устройства человека кроется вовсе не в количестве, а в качестве. Собака зарыта, таким образом, не в самом наборе генов, а в тонкой и сложной регуляции их работы.
Доктор биологических наук, биоинформатик Михаил Гельфанд заметил как-то в нашем с ним разговоре, что секвенированный геном - лишь найденный папирус, на котором виден непонятный текст. Но мы не знаем, что на нем написано, не можем перевести на свой язык. То есть мы не понимаем, как работает геном: почему в одних клетках включаются одни гены, в других клетках - иные (и благодаря этому клетки нашего организма разные), почему одни гены работают только в эмбрионе, а по мере развития человека вместо них начинают работать другие и т. д. Сами гены, как оказалось, занимают всего 1–2% длины молекулы ДНК. Напомним, что в гене содержится информация о строении белка (или нескольких белков), то есть гены кодируют белки. Но 98–99% ДНК белков не кодирует. Конечно, ученые догадывались, что не всё так просто, природа не может быть настолько расточительна и «мусорная» ДНК для чего-то очень нужна. Но, чтобы это показать, нужно было проделать много кропотливой работы, которая под силу только большому международному консорциуму. Такой масштабный замысел и стал в итоге целью проекта ENCODE (Encyclopedia of DNA Elements), в котором участвуют более 400 исследователей, члены 32 научных групп.
ДНК-энциклопедия
Работа стартовала в 2003 году. Финансировал ее Национальный институт исследований генома человека (National Human Genome Research Institute). Проект обошелся ему в 185 млн долларов США. На первом этапе, когда шла отработка методик, ученые проанализировали лишь 1% некодирующей части ДНК. С 2007 года начался второй этап, результаты которого и опубликованы одновременно в Nature, Science и других научных журналах. Но насколько велика получившаяся энциклопедия? Как пишет редактор Nature Брендан Мэйер (Brendan Maher), если распечатать все геномные данные, собранные по проекту за пять лет, при плотности 1000 пар оснований на квадратный сантиметр вышла бы распечатка 30 км в длину и 16 м в высоту. В ней содержатся 15 трлн байт информации.
По словам Мэйера, в ходе программы «Геном человека» получен «рабочий чертеж» генома. Но к этому чертежу не прилагается «руководства пользователя», и поэтому мы не знаем, как его читать. Чтобы понять это (то есть получить такой мануал), участники ENCODE стали интенсивно изучать пустые пространства между генами, предполагая, что инструкция к геному может быть записана именно там.
За пять лет они смогли перелопатить около 80% этой пустыни и нанесли на карту найденные в «мусоре» различные регуляторные участки. И чем тщательнее они исследовали ДНК, тем сложнее она оказывалась устроена - с каждым шагом вперед горы становились выше.
Транскрипционные факторы
Белки, контролирующие процесс синтеза РНК на матрице ДНК (транскрипцию) путем связывания со специфичными участками ДНК. Таким образом, они обеспечивают усиление или ослабление работы гена.
Геномная «темная материя»
Как генетики изучали ДНК, которая не делает белков? Во-первых, они убедились в том, что с некодирующих участков генома тоже образуется РНК. Основную роль в этой работе сыграла группа лаборатории КолдСпринг-Харбор, которой руководит профессор Томас Джинджерас (Thomas Gingeras).
Именно эта команда ученых доказала, что три четверти человеческой ДНК образует РНК, хотя большая часть этой РНК и не несет информации для синтеза белков клетки. Д-ру Джинджерасу и его коллегам удалось описать тысячи неизвестных ранее РНК, которые обеспечивают генную регуляцию.
Во-вторых, они пометили ДНК особым ферментом (DNaseI), который прикрепляется к определенным местам молекулы. Это и оказались регуляторные участки, которые связываются с белками - транскрипционными факторами - и через них влияют на работу генов. Одни из них расположены непосредственно рядом с генами, другие - совсем далеко от них. Доктор биологических наук Юрий Лебедев, заведующий лабораторией сравнительной функциональной геномики Института биоорганической химии им. Шемякина и Овчинникова РАН, объясняет, что задачу проекта ENCODE можно представить как «разбиение непрерывного ряда букв последовательности нуклеотидов ДНК на отдельные слова и осмысленные предложения». Для ее решения исследователи использовали не «виртуальные» биоинформатические, а прямые экспериментальные методы, например разного рода биологические чипы. «Чип можно представить как своеобразную “щетку”, каждая “ворсинка” которой представляет собой короткий кусочек ДНК с точно известным положением в геноме», - поясняет ученый. Этим методом идентифицируют положение участков ДНК, которые связываются с транскрипционным фактором. При нанесении раствора-зонда на чип на некоторых ворсинках возникает флуоресцентный сигнал.
«Если для приготовления зонда взять ДНК из разных типов клеток, можно увидеть разницу в наборе флуоресцентных сигналов, - продолжает Лебедев. - Например, в ДНК из лейкоцитов данный белок будет связываться с одними участками, а в ДНК из клеток печени или мозга спектр этих участков будет другим. Подобным же образом определяют профили метилирования геномной ДНК - одного из вездесущих клеточных механизмов, регулирующих активность генов. Чтобы найти метилированные участки ДНК, используют группу специальных метил-связывающих белков. Так получается подробная функциональная карта генома для разных типов клеток».
Метилирование ДНК
Химическая модификация молекулы ДНК без изменения состава нуклеотидной последовательности. Выражается в присоединении метильной группы (СН3--) к цитозину. От степени метилирования зависит уровень экспрессии гена. Это один из механизмов регуляции работы генов.
Дирижеры генного оркестра
Итак, на 25 тыс. генов в ДНК обнаружилось около 4 млн регуляторных участков. Каждый ген взаимодействует со множеством регуляторов. «Большинство людей представляют геном линейно - 3 млрд нуклеотидов, вытянутых в линию, - говорит Марк Герштейн (Mark Gerstein), профессор биоинформатики на отделении молекулярной биофизики и биохимии Йельского университета (США). - Но геном - трехмерный объект». По его словам, «мы будто открыли коммутационный отсек и увидели спутанный клубок проводов. И теперь пытаемся распутать этот клубок и разобраться, куда ведут провода». В статье в Nature Марк Герштейн и его коллеги пишут про сложную пространственную сеть, в которую объединены транскрипционные факторы, которые регулируют работу генов на разных уровнях: среди них есть топ-регуляторы, регуляторы среднего и низшего звена.
Ученые имели дело с большим разнообразием клеток - всего они изучили 147 типов клеток, взятых из многих тканей на разных стадиях их развития. Оказалось, что команда регуляторов, которые дирижируют генным оркестром, меняется с типом клетки и со временем. Для разных клеток исследователи создали карты активных регуляторных участков. В этом ключ к пониманию того, что клетки развиваются и проходят дифференцировку от стволовых к специализированным, и нервные клетки в итоге не похожи на мышечные. «Полученные карты генома разных клеток можно сравнить с картами, которые нам дает Google Maps, - объясняет Эрик Лэндер (Eric Lander), президент Broad Institute (объединенный институт, в состав которого входят Массачусетский технологический институт, Гарвардский университет и институт Уайтхеда). - По сравнению с ними результаты предшествующего проекта “Геном человека” давали нам взгляд на Землю из космоса. По этим снимкам невозможно определить, где проходят трассы, каков на них трафик в данное время дня, они не укажут вам на лучшие рестораны в округе или на больницы в этом городе на берегу реки». Теперь же, продолжаем сравнение, по геному стало возможно «ездить с навигатором».
Дифференцировка
Процесс специализации клетки - развития от стадии стволовой клетки до специализированной клетки какой-либо ткани.
Однонуклеотидный полиморфизм (SNP)
Точечные мутации - замена одного нуклеотида на другой. Один из наиболее распространенных вариантов генетического разнообразия.
Болезни - чаще поломка не генов, а регуляторов
В последнее десятилетие ученые активно исследуют генетическую природу различных заболеваний. Этому помогает широкогеномное генотипирование - GWAS (genome wide association study). В группе больных и в группе здоровых людей сравнивают генетическую вариабельность, или однонуклеотидный полиморфизм (SNP), - точечные мутации, выражающиеся в замене одного нуклеотида другим. Таким образом выявляют ассоциации мутаций с болезнью. Иногда удается связать болезнь с определенными генами.
Трудность состоит в том, что только 15% этих вредных мутаций приходится на гены, и тогда можно понять, что именно ломается в организме. А 85% мутаций попадает вовсе не на гены, а на ту самую межгенную «темную материю», о которой ученые до недавнего времени практически ничего не знали. «Большинство изменений, которые связаны с болезнями, лежат не в самих генах, а в переключателях», - объясняет Майкл Снайдер (Michael Snyder), исследователь-микробиолог из Стэнфордского университета (США). Поэтому работы по расшифровке генома пока дают очень малый эффект для диагностики и лечения.
«Многие исследователи находили участки человеческого генома, мутации в которых вызывают определенные болезни, - объясняет Джоб Дэккер (Job Dekker), профессор отделения биохимии и молекулярной фармакологии Медицинской школы Массачусетского университета (США). - Во многих случаях, как удалось понять, эти участки совсем не содержат генов, и тогда объяснить причину патологии трудно. Данные ENCODE показывают, что многие из этих мутаций затрагивают регуляторные элементы генов, и в некоторых случаях мы можем найти, какие гены регулируются этими элементами. Таким образом, мы можем значительно лучше понять генетические основы болезни».
Разбираясь в том, какие регуляторы ломаются и работу каких генов они нарушают, генетикам удалось обнаружить неожиданные связи между, казалось бы, весьма далекими по природе заболеваниями, пишут авторы статьи в Nature. Например, одна мутация в регуляторном участке изменяет работу нескольких генов, что может в конечном итоге привести к рассеянному склерозу, волчанке, ревматоидному артриту, болезни Крона, глютеновой болезни.
«Самый главный выход проекта - в предоставлении громадного массива данных для сравнительного анализа функциональных карт, - считает Юрий Лебедев. - Проведение такого анализа крайне важно для развития медицины, поскольку его результаты могут ответить на вопрос, что изменится в геноме больных клеток по сравнению с геномом здоровых. Если мы возьмем, скажем, ДНК из клеток двух типов, например клеток раковой опухоли легкого и неповрежденных клеток легкого (поверхностный эпителий альвеол), у них можно сравнить профиль метилирования или связь с транскрипционными факторами, и профили будут разными. Если этот результат получен не на одном пациенте, а на многих, он может стать диагностическим признаком».
Таким образом уже удалось найти мутации, которые связаны с развитием рака, - подавляющее число мутаций в раковых клетках появляются опять-таки не в генах, а в зоне «темной материи». Об этом говорит д-р Марк Рубин (Mark Rubin), специалист по генетике рака простаты из Медицинского колледжа Вейл Корнелл в Нью-Йорке. Его группа обнаружила мутации в ключевых генах, связанные с раком простаты, которые, однако, было невозможно компенсировать лекарствами. Теперь ясно, какие участки «темной материи» нарушают работу этих генов, - появились новые мишени для лекарственной терапии.
Марк Герштейн считает, что результаты проекта ENCODE найдут когда-нибудь применение в области персональной геномики: «В будущем каждый человек будет располагать собственным секвенированным геномом и сможет использовать эту информацию для получения персональной медицинской помощи. Индивидуальные генетические карты будут применяться для оценки индивидуального риска развития тех или иных болезней и для разработки индивидуальной схемы лечения».


